MALATTIE NEUROMUSCOLARI: LE NEUROPATIE |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

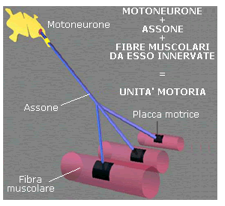 |
Ogni unità motrice comprende quattro componenti funzionali: il corpo cellulare del motoneurone, il suo assone, che decorre in un nervo periferico, la giunzione neuromuscolare e le fibre muscolari innervate dal neurone stesso. Le caratteristiche distintive di queste malattie dipendono da quale delle quattro componenti citate viene particolarmente colpita: forme che interessano principalmente i corpi cellulari (malattie del motoneurone), le fibre nervose (neuropatie periferiche) e le forme miopatiche o miopatie caratterizzate da alterazioni a livello dei muscoli.
Esistono vari criteri di classificazione dei vari tipi di neuropatia. Facendo riferimento al tipo di fibre interessate dal processo patologico si distinguono:
- neuropatie motorie
- neuropatie sensitive
- neuropatie autonomiche (anche neuropatie vegetative)
- neuropatie miste.
È anche possibile effettuare una distinzione in base al processo patologico; in questo caso si parla di:
- neuropatie demielinizzanti
- neuropatie assonali.
Nel primo caso il danno interessa la mielina, nel secondo l'assone; è opportuno precisare che, molto frequentemente, il danno è di tipo misto; sono cioè interessati sia la mielina sia l'assone.
Facendo riferimento invece al processo evolutivo si effettua la seguente classificazione:
- neuropatie acute
- neuropatie subacute
- neuropatie croniche.
Si possono anche distinguere le neuropatie facendo riferimento alla distribuzione del deficit; basandosi su questo criterio si distinguono i seguenti processi patologici:
- mononeuropatie (anche momoneuriti o mononevriti)
- multineuropatie (anche multineuriti, multinevriti o mononeuropatie multiple)
- polineuropatie (anche polineuriti o polonevriti)
- radicolopatie.
Nelle mononeuropatie il processo patologico interessa un unico tronco nervoso; nelle multineuropatie l'interessamento è relativo a più tronchi nervosi, ma il processo patologico è distinto e non contiguo; sono cioè interessati più nervi in modo asimmetrico.
Nelle polineuropatie l'interessamento è relativo a più nervi in modo contemporaneo; nelle radicolopatie invece l'interessamento è relativo alle radici dei nervi spinali.
La classificazione migliore, però, è di tipo eziologico.
Facendo riferimento alle cause è possibile suddividere le neuropatie in due grandi gruppi:
- neuropatie ereditarie
- neuropatie acquisite.
Rientrano nelle neuropatie ereditarie la HSMN (acronimo dei termini inglesi Hereditary Sensory Motor Neuropathy, neuropatia ereditaria sensitivo-motoria) - un tempo nota come malattia di Charcot-Marie-Tooth -, la neuropatia amiloidotica familiare e le neuropatie in corso di porfiria.
Le neuropatie acquisite possono riconoscere diverse cause. Molte sono associate a patologie di carattere sistemico come, per esempio, il diabete, l'insufficienza renale o le epatopatie; altre sono provocate da infezioni virali o batteriche, altre ancora sono di tipo immuno-mediato (neuropatie su base immunitaria); rientrano in quest'ultimo gruppo la sindrome di Guillain-Barré, le neuropatie vasculitiche e le neuropatie associate a gammopatie monoclonali.
Altre neuropatie di tipo acquisito sono quelle che si sviluppano in conseguenza a processi di tipo neoplastico e le neuropatie delle sindromi paraneoplastiche.
Un cenno va infine riservato alle cosiddette neuropatie iatrogene, ovvero quelle che sono causate da un processo diagnostico e/o terapeutico. Generalmente vengono distinte in due grandi gruppi: neuropatie iatrogene di tipo fisico-farmacologico e neuropatie iatrogene di tipo traumatico; fanno parte del primo gruppo quelle conseguenti a trattamenti terapeutici quali la radioterapia o le iniezioni di farmaci in un tronco nervoso; appartengono invece al secondo gruppo quelle provocate da manovre sanitarie errate o conseguenti a interventi di tipo chirurgico o sessioni a carattere diagnostico.
2. CAMPO DI APPLICAZIONE
Indicazioni relative alla gestione di pazienti con neuropatie periferiche che vengono seguiti presso l’ambulatorio di Neurologia. Di seguito verranno, pertanto, considerate alcune forme di neuropatie acquisite
3. CONTENUTO
I pazienti che accedono presso l’Ambulatorio di Neurologia per sospetto diagnostico di neuropatia saranno seguiti per la formulazione della diagnosi, qualora per essa siano sufficienti criteri minimi generali, e per follow up nel caso presentino specifiche forme di neuropatia periferica acquisite. Per le forme di neuropatia per cui si prevedano per la diagnosi criteri specifici è previsto l’invio presso reparti strutturati di neurologia.
Si ricorda che per porre diagnosi di neuropatia periferica, è necessario definire: a) criteri generali riguardanti le caratteristiche cliniche della malattia ed i dati di laboratorio, b) i criteri specifici per la diagnosi di neuropatia.
Criteri minimi generali per la diagnosi di neuropatia periferica
A – Profilo clinico:
1 – Debolezza e atrofia del muscolo;
2 – Riduzione o assenza di riflessi osteotendinei;
3 – Sintomi sensitivi o deficit specifici di modalità sensitive;
4 – Alterazioni vegetative.
B – Dati di laboratorio:
1 – Reperti elettrofisiologici (EMG e velocità di conduzione).
I criteri minimiindispensabili per la diagnosi di neuropatia periferica dovrebbero essere la positività di una voce di ciascun gruppo (Ae B) oppure le voci 1 e 3 del gruppo A, oppure la voce 1 della colonnaB.
Criteri specifici per la diagnosi di neuropatia periferica
A – Esame del liquido cerebrospinale;
B – Biopsia del muscolo e del nervo;
C – Indagine genetica familiare;
D – Analisi chimica del sangue, delle urine o dei tessuti;
E – Ricerche immunologiche;
F – Indagini radiologiche.
Di seguito saranno considerate le seguenti neuropatie acquisite:
- Polineuropatie diabetiche o da altre cause metaboliche
- CIDP (poliradicoloneuropatia demielinizzante infiammatoria cronica)
- MMN (neuropatia motoria multifocale)
- Neuropatia demielinizzante associata a paraproteine
- Polineuropatia multifocale
- Radicolopatie
- IperCKemie e disturbi neuromuscolari da alterazioni elettrolitiche
3.1 POLINEUROPATIE DIABETICHE O DA ALTRE CAUSE METABOLICHE
Il termine neuropatia metabolica include un ampio spettro di neuropatie periferiche associate a patologie sistemiche di origine metabolica. Queste patologie comprendono il diabete mellito, l’ipoglicemia, l’uremia, l’ipotiroidismo, malattie epatiche, amiloidosi, acromegalia, Porfiria, disturbi del metabolismo di lipidi e glicolipidi, deficit nutrizionali e vitaminici, disordini mitocondriali ed altro.
L’elemento comune a tutte queste malattie è l’alterazione strutturale o funzionale della porzione mielinica o assonica dei nervi periferici.
Il Diabete Mellito è la causa più comune di neuropatia metabolica seguita dall’uremia.
Le neuropatie sono caratterizzate da una progressiva perdita di funzione delle fibre nervose. Una definizione ampiamente accettata di neuropatia diabetica è la seguente: “ presenza di sintomi e/o segni di disfunzione dei nervi periferici in soggetti con diabete dopo aver escluso altre cause.”
Le neuropatie sono le più comuni complicazioni del diabete mellito, interessando più del 50% dei pazienti con DM di tipo 1 e 2. Nel DM di tipo 1, la polineuropatia distale tipicamente diviene sintomatica dopo alcuni anni di costante iperglicemia. Viceversa, pazienti con DM tipo 2 possono manifestare la polineuropatia distale dopo solo pochi anni di scarso controllo dei valori glicemici; a volte, questi pazienti hanno già la neuropatia al momento della diagnosi.
La neuropatia compromette notevolmente la qualità di vita dei pazienti.
La neuropatia diabetica puo' interessare i nervi cranici (mononeuropatie craniche), i nervi spinali (polineuropatie, neuropatie focali) o le fibre nervose del sistema nervoso autonomo.
Mononeuropatie craniche
I nervi cranici piu' frequentemente interessati sono il nervo oculomotore e il nervo facciale. L'interessamento del nervo oculomotore determina una caduta della palpebra superiore (ptosi), e una paralisi di alcuni muscoli che muovono l'occhio con conseguente visione doppia (diplopia). Se viene colpito il nervo facciale si avra' una paralisi dei muscoli facciali di un lato del volto (paralisi facciale periferica).
Polineuropatie, neuropatie focali
Il coinvolgimento dei nervi spinali è molto frequente. In genere, risultano colpiti i nervi degli arti inferiori nelle loro porzioni piu' distali, con sintomi che interessano le gambe e i piedi. La forma di piu' comune riscontro e' la "polineuropatia sensitiva distale simmetrica", che determina la comparsa di disturbi della sensibilita' (formicolii e ipoestesia) sia soggettivi che obiettivabili alla visita, accompagnati da dolori urenti che interessano le gambe e piedi.
La riduzione della sensibilita' alla pianta del piede in corso di neuropatia diabetica può predisporre alla comparsa di una temuta complicanza nota come "piede diabetico". Il disturbo di sensibilita', infatti, riduce la percezione di eventuali piccole lesioni cutanee che possono formarsi alla pianta del piede e che se non tempestivamente osservate e trattate possono determinare, lesioni ulcerative fino alla gangrena del piede. In alcuni casi ai disturbi della sensibilita' si associano deficit motori ("neuropatia sensitivo-motoria"). Piu' raramente si puo' osservare l'interessamento dei nervi intercostali con la comparsa di dolori "intercostali", spesso difficili da trattare. L'iperglicemia non controllata puo', inoltre, rendere alcuni nervi piu' suscettibili a paralisi da compressione nei punti in cui questi passano attraverso zone anatomicamente piu' strette, come per esempio al polso o al gomito (sindrome del tunnel carpale, sindrome del canale di Guyon).
Neuropatia autonomica
Si verifica nel caso siano colpite le fibre del sistema nervoso autonomo che innervano i visceri quali il cuore, l'intestino, la vescica e gli organi genitali.
I segni e i sintomi variano a seconda di quali parti del sistema nervoso autonomo sono interessate. Essi possono prevedere: capogiri e svenimenti in posizione eretta, disturbi della frequenza cardiaca, disturbi della funzione digestiva (stipsi, diarrea), della funzione vescicale e sessuale (deficit di erezione).
La gestione della neuropatia diabetica dovrebbe iniziare al momento della diagnosi del diabete. Il medico di base deve essere attento sullo sviluppo della neuropatia perché trascurare la diagnosi può determinare serie conseguenze che includono gravi disabilità ma dovrebbe dare le corrette informazioni al paziente in merito alle complicanze acute e croniche del diabete. I pazienti con neuropatia diabetica periferica richiedono frequenti follow up con particolare attenzione all’ispezione dei piedi per accentuare la necessità di aumentare la cura di se.
la diagnosi di neuropatia diabetica si fonda sul riscontro clinico di diabete, sulla presenza di segni neurologici compatibili e sulla dimostrazione di rallentamento della velocita' di conduzione nervosa dei nervi misurata attraverso l'esame elettroneurografico.
la terapia della neuropatia diabetica prevede come momento fondamentale il controllo dei valori glicemici. La normalizzazione dei livelli glicemici, infatti, puo' migliorare i sintomi. In aggiunta, si utilizzano farmaci che possano soprattutto limitare le manifestazioni dolorose associate. Tra questi i piu' utilizzati ed efficaci sono alcuni farmaci appartenenti alla classe degli antidepressivi (duloxetina, amitriptilina) e degli antiepilettici (gabapentin, pregabalin).
La neuropatia diabetica rappresenta una patologia neurologica frequente che trova le sue cause in un inadeguato controllo glicemico. Si raccomanda, pertanto, al paziente affetto da diabete di effettuare controlli periodici che permettano di individuare i segni precoci di una neuropatia diabetica cosi da evitare l'instaurarsi dei sintomi clinici e delle complicanze piu' temibili.
Nella diagnosi sono previsti alcuni esami utili ad escludere altre forme di neuropatia metabolica.
Esami utili:
- Funzionalità renale
- Funzionalità tiroidea per individuare eventuale ipotiroidismo
- Funzionalità epatica
- Dosagggio Vit B12 e Ac Folico
In caso di una neuropatia associata ad insufficienza renale e cardiaca, epatica ed edemi AAII bisognerebbe escludere l’amiloidosi procedendo ad una biopsia del grasso periombelicale da sottoporre a colorazione con rosso congo. Segni rari ma caratteristici dell'amiloidosi sono: aumento di dimensioni della lingua, comparsa di macchie di colore rosso porpora sulla pelle del volto - in particolare intorno agli occhi - e del collo.
Quando una neuropatia si manifesta con ipostenia distale ad andamento ascendente associata a dolore addominale talora molto intenso o a nausea, vomito, stitichezza, diarrea, ritenzione urinaria, incontinenza, disuria, pollachiuria, tachicardia, ipertensione, sudorazione intensa, irrequietezza, tremori bisognerebbe sospettare una porfiria, conseguente ad alterazione enzimatica congenica che implica accumulo di porfirine (pigmenti di complessa struttura che costituiscono parte di emoglobina, mioglobina, citocromi, catalasi e perossidasi),
3.2 CIDP (POLIRADICOLONEUROPATIA DEMIELINIZZANTE INFIAMMATORIA CRONICA)
La CIDP, polineuropatia demielinizzante infiammatoria cronica è una poliradicolopatia acquisita a probabile genesi immunomediata, ad andamento progressivo o recidivante in un periodo di oltre 8 settimane. Sebbene non sia chiara l’esatta patogenesi, si pensa che possa entrare in gioco contro mielina o assoni dei nervi periferici, comprese le radici dei nervi spinali, un meccanismo di tipo sia cellulare sia immunomediato. Il trattamento tempestivo della CIDP è fondamentale per prevenire la perdita assonale ed esso prevede l’uso di corticosteroidi, plasmaferesi ed immunoglobuline ev (IVIg).
La diagnosi di CIDP si basa su elementi clinici e neurofisiologici. Dal punto di vista clinico vi è un’ipostenia degli arti inferiori ed in minor misura di quelli superiori con difficoltà progressiva della deambulazione.
Dal punto di vista terapeutico negli ultimi anni diversi trials clinici hanno dimostrato l'efficacia soprattutto delle IVIg verso le quali pare esserci una buona risposta nel 60-80% delle CIDP. Tra i corticosteroidi si utilizza Prednisone alla dose di 1-1,5 mg/kg/die per 2-4 settimane. Il bolo steroideo intravenoso può essere efficace nei casi più gravi. Parecchi studi randomizzati, controllati hanno dimostrato i benefici delle IVIg nel breve termine. La dose d'attacco usuale è di 2g/kg che può essere divisa in 2-5 giorni (in media 0,4g/Kg/die per 5 gg) e la dose di mantenimento prevede la somministrazione periodica di 1g/Kg. Discreto beneficio possono trarre i pz con CIDP anche dalle plasmaferesi che possono essere utilizzate come terapia di mantenimento da sole o in associazione a terapia immunomodulatoria o immunosoppressiva. Circa il 25% dei pz non risponde a nessuno dei trattamenti su citati.
3.3 MMN (NEUROPATIA MOTORIA MULTIFOCALE)
la Neuropatia motoria multifocale (MMN) è un disordine immunomediato caratterizzato da un rallentamento progressivo, debolezza asimmetrica degli arti senza deficit sensitivi. La presentazione clinica della MMN mima quella delle malattie del secondo motoneurone, ma nello studio della conduzione nervosa dei pazienti sono stati trovati blocchi di conduzione motoria. A differenza della CIDP, il trattamento con prednisolone e plasmaferesi è generalmente inefficace nella MMN e spesso associato ad un peggioramento clinico. Solo in via teorica pare siano efficaci immunosoppressori e ciclofosfamide. Vari studi aperti e 4 trials controllati con placebo hanno evidenziato un miglioramento della forza muscolare con alte dosi di IgG Vena (in media 0,4g/Kg/die per 5 gg). Anche se studi clinici, di anatomia patologica, imaging, immunologici ed elettrofisiologici hanno migliorato le conoscenze sulla MMN in più di 15 anni, sono necessari studi ulteriori per chiarire i meccanismi patogenetici della malattia.3.4 NEUROPATIA DEMIELINIZZANTE ASSOCIATA A PARAPROTEINE
E’ sempre più frequente il riconoscimento di una polineuropatia sensitivo-motoria cronica associata ad anomalie delle immunoglobuline sieriche, anche se i confini non sono ancora del tutto ben stabiliti. L’eccesso di proteine ematiche, noto come paraproteinemia, solitamente coincide con un picco monoclonale di immunoglobuline. Si può trattare di un’anomalia isolata o rappresentare il prodotto di una neoplasia maligna delle plasmacellule, in particolare il mieloma multiplo, il plasmocitoma e la macroglobulinemia di Waldenstrom. In alcune di queste malattie si ha evidenza della presenza di un anticorpo patogeneticamente attivo, diretto contro componenti della mielina o dell’assone. Forme particolari di neuropatia si associano anche ad amiloidosi. Bisogna infine considerare i casi di neuropatia associata a proteina monoclonale IgM non neoplastica che sono molto più comuni rispetto a quelli associati a malattie maligne plasmacellulari. La polineuropatia associata a gammopatia monoclonare colpisce prevalentemente gli uomini nella sesta-settima decade di vita. L’esordio è insidioso ed è caratterizzato da sintomi prevalentemente sensitivi con ipotrofia muscolare lieve ed abolizione dei ROT nella fase avanzata di malattia. La maggior parte dei casi presenta un quadro elettromiografico demielinizzante o misto. Nonostante le IgG siano le paraproteine più frequenti negli adulti, la polineuropatia si associa più frequentemente alla classe IgM. Diversi studi hanno dimostrato che il sottogruppo con paraproteina IgM manifesta più spesso gravi alterazioni sensitive ed alterazioni elettromiografiche in senso demielinizzante, rispetto al gruppo con IgG. Tra le paraproteine gli anticorpi più rappresentati sono gli anti MAG (anti glicoproteine associate alla mielina) responsabili di una forma prevalentemente sensitiva caratterizzata da difficoltà di deambulazione, atassia, tremore e Romberg positivo e gli anti GM1 responsabili di blocchi di conduzione motoria multifocali. Solo una piccola percentuale di pz con anti MAG o GM1 tende a sviluppare un mieloma o una malattia di Waldenstrom. Dal punto di vista terapeutico si sono dimostrate efficaci IGEV, terapia immunosoppressiva con ciclofosfamide o fludarabina, micofenolato ev o clorambucile per os.
3.5 POLINEUROPATIE MULTIFOCALI E ASIMMETRICHE (MONONEUROPATIE O MONONEURITI MULTIPLE)
Alcune condizioni sistemiche, soprattutto le vasculiti, possono accompagnarsi ad un interessamento acuto o subacuto di molteplici nervi singoli in successione o in modo quasi contemporaneo. Il quadro che ne risulta è denominato mononeurite multipla. Gli esempi più evidenti di questa sindrome si associano a vasculiti quali poliartrite nodosa, vasculite isolata dei nervi periferici, granulomatosi di Wegener, crioglobulinemia, HIV e più raramente a LES ed artrite reumatoide. Le caratteristiche distintive sono rappresentate da un’evoluzione acuta o sub-acuta di una paralisi sensitivo-motoria completa o pressoché completa di un singolo nervo periferico, con interessamento asimmetrico, senza chiara distribuzione di sede o di dolore prossimale o distale. Nel sospetto di vasculite, a scopo diagnostico possono essere utili indagini ematiche mirate quali ricerca di anticorpi pANCA specifici della poliartrite nodosa, di anticorpi c-ANCA specifici della sindrome di Churg-Strauss e dell’arterite di Wegner, di FR specifico dell’artrite reumatoide. Certamente dirimenti possono essere la biopsia dei nervi che nella poliartrite nodosa può evidenziare flogosi perivascolare e necrosi. Dal punto di vista terapeutico efficaci si sono dimostrate alte dosi d’attacco di corticosteroidi seguite da una più bassa posologia associata a terapia immunosoppressiva a base di Ciclofosfamide e Azatioprina. Ad esempio per la Poliartrite Nodosa: Metilprednisolone 1.5 mg/kg per diversi giorni seguito da trattamento orale associato a Ciclofosfamide o Azatioprina.
Dal punto di vista procedurale, quando viene posto il sospetto diagnostico di questa tipologia di patologia il passo successivo è quello di favorire la prosecuzione dell’iter diagnostico terapeutico presso ambulatori dedicati di ematologia o reumatologia a seconda dei casi.
3.6 RADICOLOPATIE
Si tratta del gruppo di patologie clinicamente più complesse nell’ambito del sistema nervoso periferico. L’interessamento di radici spinali multiple si manifesta con una costellazione di sintomi e segni distintivi diversi da quelli delle polineuropatie e delle mononeuropatie multiple. L’ipostenia causata dalla poliradiculopatia è caratteristicamente asimmetrica e variamente distribuita nelle porzioni prossimali e distali degli arti, riflettendo il fatto che i muscoli colpiti hanno la medesima innervazione radicolare. Tuttavia, i muscoli con la stessa innervazione non sono necessariamente interessati nello stesso modo, in conseguenza del contributo non uniforme di una data radice a ciascun muscolo. Il deficit sensitivo tende ad essere, analogamente, distribuito ad aree distinte ed interessa sia le porzioni prossimali che quelle distali di un dermatomero. Di regola il dolore, non costante, è comune, mentre le alterazioni sensitive tendono ad essere meno pronunciate rispetto a quelle motorie. In accordo con un quadro di interessamento radicolare, alcuni riflessi possono essere risparmiati. La normalità del riflesso achilleo e l’assenza del rotuleo o viceversa sono particolarmente suggestivi di poliradiculopatia. Il dolore associato alle affezioni delle radici nervose è comune, ma non costante, e assume la forma di una stilettata proiettata nella regione innervata dalla radice interessata.Le patologie che interessano le radici nervose possono essere raggruppate in tre ampie categorie: 1) patologie della colonna che comprimono le radici; 2) patologie infiltrative delle meningi che coinvolgono secondariamente le radici nel punto in cui attraversano lo spazio subaracnoideo, compresa l’infiltrazione granulomatosa neoplastica; 3) le neuropatie primitive, solitamente infiammatorie, infettive o diabetiche, che hanno una predilezione per la porzione radicolare dei nervi.
Dal punto di vista diagnostico utili: EMG con presenza di potenziali sensitivi a livello dei nervi che si distribuiscono alle aree di deficit sensitivo e innervano i muscoli ipostenici e denervati ad indicare che la lesione si localizza prossimamente ai gangli delle radici dorsali e risparmia gli assoni sensitivi periferici e con perdita delle risposte tardive F ed H; RMN spinale che può evidenziare anomalie del canale con effetto sulle radici ed Esame liquorale dove si può riscontrare aumentata concentrazione proteica e pleiocitosi. Dal punto di vista terapeutico necessaria, una volta fatta la diagnosi, la rimozione della causa.
3.7 IPERCKEMIE E DISTURBI NEUROMUSCOLARI DA ALTERAZIONI ELETTROLITICHE
Fibre nervose e muscolari mantengono un ambiente interno fluido diverso dall’ambiente esterno e dallo spazio extracellulare. I principali costituenti intracellulari solo: K+; Mg+ e P-, i principali costituenti extracellulari sono: Na+; Ca++ e Cl-. La permeabilità selettiva delle membrane cellulari e la pompa del Na+ mantengono un equilibrio elettrico chiamato Potenziale di Riposo. Alla base di questo potenziale vi sono il K+ (che è 30 volte più concentrato all’interno che non all’esterno) ed il Na+ (10-12 volte più concentrato all’esterno). L’interno della cellula ha una carica negativa, il K+ tende ad uscire secondo gradiente di concentrazione ma viene trattenuto all’interno secondo gradiente elettrico. Stessa cosa vale per il Na+. Normalmente il potenziale di membrana è -90uV, dopo una scarica il Na+ entra nella cellula e depolarizza la membrana fino a + 40 uV inducendo il Potenziale d’Azione. Progressivamente, però, si riduce l’ingresso di Na+ e aumenta l’efflusso di k così che il potenziale di membrana tende a tornare ai valori iniziali. Una volta che si è creato un Potenziale d’azione esso diffonde per tutta la fibra secondo un criterio di “tutto o nulla”. La regolare trasmissione degli impulsi lungo le fibre nervose è elemento necessario per una regolare attività muscolare. Una fibra muscolare, infatti, per l’avvio di una contrazione dipende da uno specifico impulso nervoso. In caso di problemi nella conduzione degli impulsi per alterazioni elettrolitiche possono registrarsi scosse muscolari, spasmi e crampi. Se la concentrazione del K+ scende sotto i 2.5 mEq/l o sale sopra i 7 mEq/l si ha perdita di forza a livello del tronco e delle estremità. Sotto i 2 mEq/l e sopra i 9 si instaura una paralisi flaccida che finisce per coinvolgere la muscolatura respiratoria. Stessa cosa può manifestarsi in caso di ipofasfatemia. Debolezza muscolare associata a spasmi tetanici e crisi convulsive può aversi in caso di basse concentrazioni di Mg+. Per il resto alterazioni nella concentrazione di Na+ e Ca++ sono responsabili più che di problemi neuromuscolari, di alterazioni della coscienza e crisi convulsive.
Ovviamente non sono solo le alterazioni elettrolitiche ad indurre disfunzioni muscolari ma anche malattie proprie dei muscoli. In tutte le malattie che provocano lesioni estese delle fibre muscolari striate gli enzimi muscolari fuoriescono dalle fibre nel torrente circolatorio. Quelli che si è soliti determinare di routine sono essenzialmente le CK e le LDH ed in particolare le CK MM.
Nei pazienti con distruzioni massive del muscolo, i valori plasmatici di CK superano spesso le 1000 unità e possono raggiungere anche le 40000 unità o più. Ovviamente le cause di iperCkemia sono diverse e non tutte sono associate alla distruzione del muscolo
Incrementi di CK possono essere registrati in:
- femmina portatrice sana della forma pseudoipertrofica di Duchenne
- pazienti con atrofia muscolare spinale e con SLA
- maschi afro-americani (iperCKemia idiopatica)
- ipotiroidismo ed alcolismo
- miopatie tossiche, per es quelle causate dall’uso di farmaci ipolipemizzanti
- difetti muscolari ereditari es distrofia di Becker caratterizzata da debolezza prossimale ed ipertrofia dei polpacci
- Miopatie ereditarie da deficit di carnicina-palmitoil trasferasi, carenza di mioadenilato deaminasi, glicogenosi e alcune forme di miopatie a corpi inclusi.
- Miopatia di Miyoshi, miopatia distale che insorge in giovani adulti portatori di un deficit di disferlina. Questa forma può anche presentarsi tra i 15 e i 25 anni come una forma asintomatica ma con un consistente aumento di CK; in questo caso l’iniziale rialzo delle CK è seguito da una progressiva comparsa di debolezza distale.
- Mutazioni del gene della caveolina-3 e della titina.
Se tutte le altre cause di elevati valori plasmatici di CK che persistono per mesi o anni sono state escluse, in particolar modo quelle dovute ad un esercizio fisico o a farmaci che provocano delle lesioni muscolari, è opportuno seguire il paziente nel tempo con lo scopo di individuare l’insorgenza di un’eventuale debolezza di modesta entità, o eseguire una biopsia muscolare con l’intento di porre una diagnosi di distrofia muscolare in fase precoce.
La misurazione di LDH e varie transaminasi sieriche non è particolarmente utile per la diagnosi di malattia muscolare a causa della distribuzione ubiquitaria di questi enzimi in molti tessuti. Ovviamente un aumento inspiegabile di tutti gli enzimi muscolari (CK, LDH, SGOT ecc) deve far pensare ad un trauma anche qualora esso non sia evidente.
4. INDAGINI DI LABORATORIO E STRUMENTALI NECESSARIE ALLA DIAGNOSI
4.1 Esami di laboratorio
- Funzionalità renale
- Funzionalità tiroidea per individuare eventuale ipotiroidismo
- Funzionalità epatica
- Dosaggio Vit B12, Ac Folico, CK e LDH, Na+; K+; Mg; Ca++; Ph-
- In caso di caso di una neuropatia associata ad insufficienza renale e cardiaca, epatica ed edemi AAII escludere Amiloidosi con biopsia del grasso periombelicale sottoposto a colorazione con rosso congo.
- In caso di neuropatia ad andamento ascendente associata ad intenso dolore addominale ed eventuali nausea, vomito, stitichezza, diarrea, ritenzione urinaria, incontinenza, disuria, pollachiuria, tachicardia, ipertensione, sudorazione intensa, irrequietezza, tremori escludere Porfiria conseguente ad alterazione enzimatica congenica che implica accumulo di porfirine (pigmenti di complessa struttura che costituiscono parte di emoglobina, mioglobina, citocromi, catalasi e perossidasi),
- Elettroforesi delle proteine ed in caso di riscontro di IgM o IgG approfondire l’indagine con anticorpi anti MAG per neuropatie prevalentemente sensitive o anti GM1 per neuropatie prevalentemente motorie.
- In caso di mononeurite multiple utili ricerca di: anticorpi pANCA specifici della poliartrite nodosa, anticorpi c-ANCA nella sindrome di Churg-Strauss e arterite di Wegner, FR nella artrite reumatoide.
4.2 EMG e velocità di conduzione
Lo studio della funzione nervosa periferica prevede la stimolazione transcutanea dei nervi motori e sensitivi e la registrazione del relativo potenziale d’azione del muscolo (cMAP) e del potenziale d’azione del n. sensitivo (sNAP). Gli studi sulla conduzione nervosa motoria e sensitiva espressi in termini di: ampiezza, velocità di conduzione e latenze distali, consentono di ottenere
informazioni quantitative e qualitative sulla morfologia dell’onda e sulla dispersione degli impulsi.
Tecniche per lo studio della velocità di conduzione
Si stimola un nervo accessibile con un elettrodo di superficie attraverso la cute. Il Potenziale d’Azione che ne deriva viene registrato da elettrodi posti sulla cute:
- sopra il ventre muscolare, distalmente, in caso di stimolazioni di fibre motorie di un nervo motore o misto
- al di sopra del nervo in sede più prossimale, nel caso di tecniche ortodromiche delle fibre sensitive di grande diametro stimolate a livello dei nervi digitali.
- Al disopra del nervo in sede distale qualora si utilizzi la tecnica antidromica per lo studio della conduzione delle fibre sensitive
- Al di sopra del nervo, in sede più prossimale, per studi sulla conduzione di un nervo misto
- Con elettrodi ad ago è possibile registrare i potenziali d’azione mano a mano che questi diffondono lungo il nervo.
I tempi di conduzione dall’elettrodo di stimolazione più distale al punto di registrazione vengono denominati: Latenza Distale e Latenza di Picco. Se sia applica un secondo stimolo in un nervo misto più prossimale è possibile misurare un secondo tempo di conduzione su un segmento di nervo più lungo.
La Velocità di Conduzione massima si ottiene dividendo la distanza (in mm) tra i due punti di stimolazione nelle fibre motorie o fra i due punti di registrazione nelle fibre sensitive. Per la differenza tra i tempi di conduzione (in msec). Questa velocità indica la velocità di propagazione delle fibre di maggiore diametro. La Velocità di Conduzione max varia da un minimo di 40-45 m/sec a un massimo di 65-75 m/s a seconda del n indagato (più basso nelle gambe rispetto alle braccia). La velocità si riduce con il freddo.
 |
 |
Sono stati stabiliti anche i valori di riferimento per le latenze distali dalle sedi più lontane di stimolazione di vari nervi misti ai rispettivi muscoli, ad es se si stimola il n mediano al polso la latenza di conduzione motoria attraverso il tunnel carpale ai muscoli dell’eminenza tenar innervati dal n mediano è sempre < 4.5
Nelle neuropatie assonali (neuropatia alcolico-nutrizionale, carcinomatosa, uremica, diabetica ecc) la velocità di conduzione varia da valori ai limiti inferiori della norma a valori mediamente rallentati, mentre sono diminuite le ampiezze dei Potenziali dei nervi sia motori sia sensitivi. Le neuropatie demielinizzanti di tipo acuto (Guillain Barré) o di tipo cronico (infiammazione cronica, leucodistrofia metacromatica, malattia di Krabbe, malattia di Charcot- Marie-Tooth) presentano invece marcata riduzione della velocità di conduzione e della latenza distale.
Potenziali d’azione sensitivi
Quando vengono stimolate le fibre motorie di un nervo misto, si può facilmente registrare un CMAP di molte centinaia di microvolt con elettrodi posti sulla cute che ricopre il muscolo. Quando si cerca di misurare i potenziali sensitivi, si dovrà registrare l’attività delle fibre nervose stesse. Viene così a mancare l’amplificazione fornita da tutte le fibre muscolari di molte unità motorie. Quindi è necessaria un’amplificazione elettronica sebbene comunque la velocità di conduzione sensitiva può essere difficile da registrare anche quando vengono usate sofisticate tecniche di averaging.
Valori di riferimento
|
Studi di conduzione nervosa motoria |
|||||||
Nervo |
Sede di stimolazione distale |
Altre sedi di stimolazione |
Sede registrazione |
Latenza distale (ms) |
Ampiezza (mV) |
VC (m/sec) |
Distanza cm |
Latenza onda F (ms) |
Mediano |
polso |
gomito |
APB (6) |
< 4.2 |
> 4.4 |
> 49 |
6-8 |
< 31 |
Ulnare |
polso |
Bg(1), AG(2) |
ADM (7) |
< 3.4 |
> 6 |
> 49 |
5.5-7.5 |
< 32 |
Radiale |
avambraccio |
gomito, SG (3) |
EIP (8) |
< 5.2 |
> 4 |
> 50 |
10 |
NA |
Peroneale |
caviglia |
BFH*, AFH (4) |
EDB (9) |
< 5.8 |
> 2 |
> 42 |
6-11 |
< 58 |
Peroneale |
BFH* |
AFH (4) |
TA |
< 3 |
> 5 |
> 42 |
10 |
NA |
Tibiale |
caviglia |
PF (5) |
AH (10) |
< 6.5 |
> 3 |
> 41 |
6-8 |
< 59 |
* BFH: sotto il capitello, (1) BG: ulnare sotto il gomito; (2) AG: ulnare sopra il gomito; (3) SG: solco spirale, (4) AFH: sopra il capitello; (5) PF: cavo popliteo; (6): APB: abduttore breve pollice; (7) ADM: adduttore V dito; (8) EIP: estensore proprio dell’indice; (9) EDB: estensore breve delle dita; (10) AH: abduttore alluce
|
Studi di conduzione nervosa sensitiva |
||||||
Nervo |
Sede di stimolazione distale |
Sede registrazione |
Latenza (ms) |
Latenza al picco (ms) |
Ampiezza (mV) |
VC (m/sec) |
Distanza cm |
Mediano |
polso |
2° dito |
< 2.5 |
> 3.5 |
> 20 |
> 50 |
13 |
Ulnare |
polso |
5° dito |
< 2.1 |
> 3 |
> 15 |
> 52 |
11 |
Radiale |
avambraccio |
polso |
< 1.9 |
< 2.8 |
> 20 |
> 48 |
10 |
Surale |
polpaccio |
caviglia |
< 3.2 |
< 4.4 |
> 6 |
> 42 |
14 |
4.2a Blocco di conduzione
Stimolando un nervo motore in più punti lungo il suo decorso, è possibile evidenziare alcuni tratti nei quali la conduzione è parzialmente “bloccata” o rallentata. Da questi dati si evince la presenza di processi demielinizzanti multifocali dei nervi motori. Tecnicamente la presenza di blocchi di conduzione è dimostrata da una riduzione dell’ampiezza del CMAP elicitato da una stimolazione prossimale lungo il nervo motore rispetto alla stimolazione in una zona distale. Blocchi di conduzione si hanno in diverse neuropatie demielinizzanti acquisite su base immunitaria tra cui: Guillain Barré, Neuropatia demielinizzante infiammatoria cronica, blocco di conduzione multifocale legato ad anticorpi anti GM1.
La compressione focale di un tronco nervoso, come nelle sindromi da intrappolamento, può provocare un rallentamento localizzato o un blocco di conduzione, forse per demielinizzazione segmentaria nella sede dello schiacciamento. La dimostrazione di queste alterazioni localizzate della conduzione conferma l’ipotesi di un intrappolamento del tronco nervoso. Ad es. se la latenza distale del n mediano è > 4.5 ms mentre quella del n ulnare rimane normale è probabile che vi sia una compressione del n mediano a livello del tunnel carpale. Un simile rallentamento focale o un blocco parziale di conduzione può essere registrato nel n ulnare al gomito e nel n peroneale in corrispondenza della testa della fibula.
4.2b Studi elettrodiagnostici delle radici nervose e dei segmenti spinali
Riflesso H
Lo studio del riflesso H e dell'onda F fornisce informazioni sulla conduzione degli impulsi attraverso i segmenti prossimali dei nervi. Nel 1918 Hoffman dimostro' che la stimolazione sottomassimale dei nervi misti sensitivo-motori, non sufficienti a produrre una risposta diretta motoria, induce una contrazione muscolare (onda H) dopo una latenza assai superiore alla risposta motoria diretta. Questo riflesso deriva dall'attivazione delle fibre afferenti dai muscoli fusiformi (gli stessi assoni che conducono la scarica afferente del riflesso osteotentineo) e la lunga latenza rappresenta il tempo necessario agli impulsi per raggiungere il midollo spinale attraverso le fibre sensitive, per effettuare la trasmissione sinaptica con le corna anteriori e per essere condotti lungo le fibre motorie fino al muscolo. Lo studio del riflesso H è utile nelle radicolopatie soprattutto L5-S1.Piu' del riflesso H per le radicolopatie si studia l'onda F. Essa è evocata da uno stimolo sovramassimale di un nervo SM. Dopo una latenza maggiore della risposta motoria diretta (latenza di 28-32 ms negli arti superiori e di 40-50 ms negli arti inferiori), si registra un secondo piccolo potenziale d'azione muscolare (onda F).L'onda F è prodotta dagli impulsi che viaggiano in modo antidromico lungo le fibre motorie fino alle cellule delle corna anteriori, una piccola parte delle quali può essere attivata e produrre una risposta ortodromica che viene registratat in un muscolo distale. Lo studio della risposta F fornisce più informazioni rispetto allo studio dell'onda H sulla conduzione del nervo prossimale e della radiche in quando l'onda F attraversa solo le radici ventrali e può essere evocata da più muscoli. Un'onda F normale e un riflesso H assente è indice di una patologia dei nervi e delle radici sensitive.
Blink Reflex
Utile per le patologie demielinizzanti e di altro genere di V e VII nc
Il n sovraorbitario viene stimolato per via transcutanea e la chiusura di entrambi i m orbicolari viene registrata con elettrodi di superficie
R1: 10 msec dopo l’applicazione dello stimolo
R2: 30 msec dopo e 35 msec controlateralmente
L’evocazione del Blink Reflex riflette l’integrità del circuito costituito da afferente del n trigemino, dalle efferente del n facciale, dagli interneuroni del ponte (R1) e dalla parte caudale del bulbo (R2)
R1 e R2 sono ritardate nella paralisi di Bell solo dal lato affetto.
Voluminosi neurinomi del n acustico possono interferire con la porzione trigeminale della via riflessa e determinare una risposta patologica dal lato affetto. Le risposte possono essere inconsistenti nelle patologie del tronco.
Esame ad ago del muscolo
Elettrodi monopolari o concentrici ad ago vengono inseriti nel ventre muscolare e registrano l’attività che deriva dalla contrazione. La scarica di una singola fibra determina un potenziale trifasico
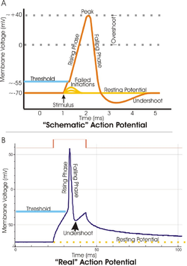 |
L’attività elettrica dei muscoli si registra sia a riposo che durante la contrazione attiva. Normalmente le fibre non scaricano spontaneamente ma solo se sono attivate ed in genere la loro attività è simultanea a quella di tutte le altre che compongono la medesima unità motoria.
Generalmente il potenziale di unità motoria è sempre trifasico ma può avere anche più fasi.
In realtà una fibra muscolare è dotata di attività tonica che viene registrata come potenziale negativo della durata di 0.5-1 msec di piccola ampiezza che è il potenziale di placca in miniatura. Quando si inserisce un elettrodo in prossimità di una placca viene registrata una serie di scariche ad elevata frequenza di potenziali bifasici di attività tra 100-300 uV: si tratta di potenziali di placca.
In sintesi, quando si inserisce nel ventre muscolare un elettrodo di registrazione ad ago vengono registrati:
MEPP: potenziali di placca in miniatura
PP: potenziali di placca
Se si fa contrarre il muscolo si osserva il reclutamento progressivo di sempre più unità motorie fino ad un “pattern di interferenza completo”
 |
 |
Il PP è dovuto all’inserzione dell’ago in prossimità della placca. Vi è un inizio di reclutamento in corso di contrazione volontaria che induce un pattern di interferenza completo.
Se un muscolo è indebolito per un processo di denervazione o se la conduzione elettrica è bloccata vi sarà un n minore di unità motorie che scaricano a frequenze da moderatamente rapide a molto rapide. Invece per sforzo volontario scarso e di lesione del motoneurone superiore i potenziali di unità motoria scaricano in n ridotto, a frequenza più bassa ed in modo irregolare.
![]()
| Biografia |
- Classification and diagnosis of the inherited neuropathies, Mary M. Reilly Ann Indian Acad Neurol. 2009 Apr-Jun; 12(2): 80–88.
- Pressure-relieving interventions for treating diabetic foot ulcers. Lewis J, Lipp A.Cochrane Database Syst Rev. 2013 Jan 31;1:CD002302. doi:
- Diabetic somatic neuropathy.Kamenov ZA, Traykov LD. Adv Exp Med Biol. 2012;771:155-75. Review.
- Diabetic autonomic neuropathy.Kamenov ZA, Traykov LD.Adv Exp Med Biol. 2012;771:176-93. Review.
- Insulin Neuritis and Diabetic Cachectic Neuropathy: a review. Knopp M, Srikantha M, Rajabally YA. Curr Diabetes Rev. 2013 Mar 14. [Epub ahead of print]
- Chronic inflammatory demyelinating polyradiculoneuropathy: diagnostic and therapeutic challenges for a treatable condition. Jean-Michel Vallat, Claudia Sommer, Laurent Magy . The Lancet Neurology, Volume 9, Issue 4, Pages 402 - 412, April 2010
- Multifocal motor neuropathy, Jan-Thies H Van Asseldonk MD a, Hessel Franssen PhD a , Renske M Van den Berg-Vos PhD b, John HJ Wokke PhD b , Dr Leonard H Van den Berg The Lancet Neurology, Volume 4, Issue 5, Pages 309 - 319, May 2005
- Systematic reviews of treatment for inflammatory demyelinating neuropathy- R. A. C. Hughes Journal of Anatomy Volume 200, Issue 4, pages 331–339, April 2002
- Standard and escalating treatment of chronic inflammatory demyelinating poliradiculoneuropathy . Yoon M-S; Chan A; Gold R, Therapeutic Advances in Neurological Disorders, 4(3) 193-200 (2011)
- European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of paraproteinemic demyelinating neuropathies. Report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society. Joint Task Force of the EFNS and the PNS. Journal of the Peripheral Nervous System 11:9-19 (2006)
- Paraproteinaemic demyelinating neuropathies. Hadden R.D.M et al, Charter 22, European Handbook of Neurological Management: Volume 1, 2nd Edition 2011
- Principi di Neurologia - Adams e Victor – VIII ottava edizione